
Thomas D. Schneider, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Building 558, Room 5
- Frederick, MD 21702-1201
- 301-846-5581
- schneidt@mail.nih.gov
RESEARCH SUMMARY
Dr. Schneider is interested in discovering and exploring the fundamental mathematics of biology: "Living things are too beautiful for there not to be a mathematics that describes them." He uses the mathematics of information theory, first developed by Claude Shannon in 1948.
Dr. Schneider first discovered that binding sites on nucleic acids usually contain just about the amount of information needed for molecules to find the sites in the genome. Information is measured in bits, the choice between two equally likely possibilities. It is the number of times one needs to divide the possibilities to reach a subset of objects. That is, the log base 2 of the number of posibilities is the number of bits. For example, ribosome binding sites in E. coli have about 10 bits of information per site on the average. To find the roughly 4000 gene starts in the 4 million base E. coli requires about log2(4,000,000/4,000) = 10 bits, close to the information measured in the ribosome binding sites.
Schneider and then high-school student Mike Stephens invented sequence logos to understand the patterns at donor and acceptor human RNA splice junctions. Sequence logos are now widely used in molecular biology.
The relationship between information, measured in bits, and the binding energy is a fundamental problem in biology. The Second Law of Thermodynamics gives the ideal relationship for converting the energy dissipated during molecular binding to bits. Using this conversion factor Dr. Schneider discovered that binding sites are 70% efficient. It turns out that rhodopsin in the eye and muscle are also 70% efficient. Dr. Schneider has discovered the basic mathematics that gives this general result.
For more information, see https://alum.mit.edu/www/toms/
For current publications, see the Google Scholar list: https://scholar.google.com/citations?hl=en&user=1p-4Z14AAAAJ&sortby=pubdate
Areas of Expertise
Research
Information Theory in Molecular Biology
![]()
Sequence logos were invented by Tom and Mike Stephens.
Shannon's measure of information is useful for characterizing the DNA and RNA patterns that define genetic control systems. Dr. Schneider has shown that binding sites on nucleic acids usually contain just about the amount of information needed for molecules to find the sites in the genome. This is a working hypothesis, and exceptions can either destroy the hypothesis or reveal new phenomena. For this reason, he is studying several interesting anomalies.
The first major anomaly was found at bacteriophage T7 promoters. These sequences conserve twice as much information as the polymerase requires to locate them. The most likely explanation is that a second protein binds to the DNA. In another case, he discovered that the F incD region has a three-fold excess conservation, which implies that three proteins bind there. Both anomalies are being investigated experimentally. Thus, the project has three major components: theory, computer analysis, and molecular biology experiments. The theoretical work can be divided into several levels. Level 0 is the study of genetic sequences bound by proteins or other macromolecules, briefly described above. The success of this theory suggested that other work of Shannon should also apply to molecular biology. Level 1 theory introduces the more general concept of the molecular machine, and the concept of a machine capacity equivalent to Shannon's channel capacity. In Level 2, the Second Law of Thermodynamics is connected to the capacity theorem, and the limits on the functioning of Maxwell's Demon become clear. The practical application of this work for most molecular biologists will be the replacement of consensus sequences with better models. Consensus sequences are being used to characterize the binding sites of macromolecules on DNA and RNA. After aligning a set of binding-site sequences, the most frequent base is chosen. A position that contains 100 percent As will be represented by an A, while a position that is only 75 percent A will also be represented by an A. The consensus is frequently used to search for binding sites, and the number of mismatches to the consensus is counted. A mismatch to a 100 percent A position is much more severe than one to a 75 percent A, but this is not accounted for so the researcher is misled. Mathematically robust graphical replacements for the consensus sequences called the Sequence Logo and Sequence Walkers won't discard hard-earned data. The Walker, which is patented, has direct medical application because it can be used to distinguish polymorphisms from mutations in human sequences. The Walker method allows one to display many different binding sites simultaneously. This bird's-eye-view is a powerful tool for gene structure analysis. We collaborated on this research with Peter Rogan, Allegheny University of the Health Sciences and many other people.
My most recently published discovery is that many molecular machines are 70% efficient. This result is explained using high dimensional geometry.
Nanotechnology
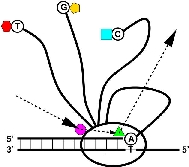
Molecular information theory tells us how molecules function. The theory therefore tells us how to build useful devices at the molecular level. We have several projects in nanotechnology. Nanoprobes are single-molecule detectors while the Medusa(TM) Sequencer is a single-molecule DNA sequencing device.
Further information may be found on the web at:
Publications
- Bibliography Link
- View Dr. Schneider's Complete Bibliography at Google Scholar
Information content of binding sites on nucleotide sequences
Sequence Logos: A New Way to Display Consensus Sequences
Sequence Walkers: a graphical method to display how binding proteins interact with {DNA} or {RNA} sequences
70% efficiency of bistate molecular machines explained by information theory, high dimensional geometry and evolutionary convergence
A brief review of molecular information theory
Biography

Thomas D. Schneider, Ph.D.
After receiving a B.S. in biology at the Massachusetts Institute of Technology, Dr. Schneider obtained his Ph.D. in molecular biology with Dr. Larry Gold in the Department of Molecular, Cellular and Developmental Biology at the University of Colorado, Boulder. His thesis and postdoctoral work, also done at Boulder, were on the application of information theory to nucleic-acid binding sites. He helped to organize and run the international news group bionet.info-theory, which is devoted to the application of information theory to biology. He was a member of the GenBank Advisory Committee and is a member of the American Association for the Advancement of Science (AAAS), The Scientific Research Society Sigma Xi and the Institute of Electrical and Electronics Engineers (IEEE) Information Theory Society.
Covers

Comparative analysis of tandem T7-like promoter containing regions in enterobacterial genomes reveals a novel group of genetic islands
Twelve prophage-like T7 islands have been discovered in pathogenic bacterial genomes. These islands contain two or three tandem T7-like promoters that should be activated when a bacterial cell is infected by bacteriophage T7 or a related phage. The illustration shows genetic maps for four of the islands, Ty2, BS512, E22 and ECA, which are found in the genomes of S. enterica Ty2, S. boydii BS512, E. coli E22 and E. carotovora SCRI1043 respectively. The T7-like promoters are represented by different colored bent arrows (red, T7; green, K1F; cyan, T3; magenta, unknown T7-like) and by corresponding sequence walkers. As in previously known mobile genetic elements, two of the islands, Ty2 and BS512, are adjacent to a tRNA-Gly gene (pink arrows) and have direct repeats of the 3' end of the tRNA gene (pink arrow tips). The other two islands, E22 and ECA, have different direct repeats on their ends (cyan chevron arrows). Each island encodes an integrase (blue arrows), several putative phage-related proteins (other arrows) and often several insertion sequence elements (white arrows).
Chen.Schneider-island2006 Z. Chen and T. D. Schneider. Nucleic Acids Res. 34:1133-1147, 2006.

Strong minor groove base conservation in sequence logos implies DNA distortion or base flipping during replication and transcription initiation
Dubbed "Tom's T" by Dhruba Chattoraj, the unusually conserved thymine at position +7 in bacteriophage P1 plasmid RepA DNA binding sites rises above repressor and acceptor sequence logos. The T appears to represent base flipping prior to helix opening in this DNA replication initation protein.
Schneider.baseflip.2001 T. D. Schneider. Nucleic Acids Res. 29:4881-4891, 2001.
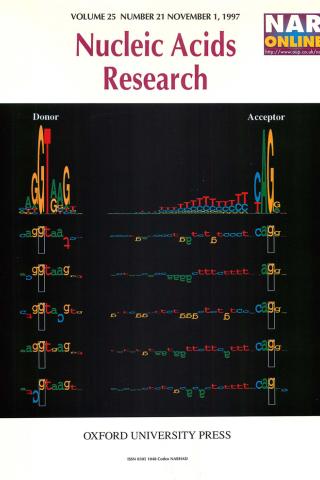
Sequence walkers: a graphical method to display how binding proteins interact with DNA or RNA sequences
A graphical method is presented for displaying how binding proteins and other macromolecules interact with individual bases of nucleotide sequences. Characters representing the sequence are either oriented normally and placed above a line indicating favorable contact, or upside-down and placed below the line indicating unfavorable contact. The positive or negative height of each letter shows the contribution of that base to the average sequence conservation of the binding site, as represented by a sequence logo. These sequence 'walkers' can be stepped along raw sequence data to visually search for binding sites. Many walkers, for the same or different proteins, can be simultaneously placed next to a sequence to create a quantitative map of a complex genetic region. One can alter the sequence to quantitatively engineer binding sites. Database anomalies can be visualized by placing a walker at the recorded positions of a binding molecule and by comparing this to locations found by scanning the nearby sequences. The sequence can also be altered to predict whether a change is a polymorphism or a mutation for the recognizer being modeled.
T. D. Schneider. Nucleic Acids Res. 1997 Nov 1;25(21):4408-15.
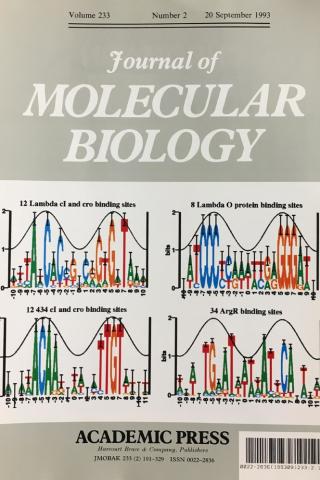
Information analysis of sequences that bind the replication initiator RepA
The tall letters represent the highly conserved bases in DNA binding sites of several prokaryotic repressors and activators. Conservation is strongest where major grooves of the double helical DNA (represented by crests of a cosine wave) face the protein. This shows that conservation analysis alone can be used to predict the face of DNA that contacts the proteins.
Papp.Schneider1993 P. P. Papp D. K. Chattoraj, and T. D. Schneider. J. Mol. Biol. 233:219-230, 1993.
Resources
See: https://alum.mit.edu/www/toms/#Tools